

MOLECULAR CHARACTERIZATION OF
HEMOPHILIA A IN SOUTHEAST BULGARIA
Sukarova Stefanovska E1, Tchakarova P2, Petkov GH2, Efremov GD1,*
*Corresponding Author: Professor Dr. Georgi D. Efremov, Research Centre for Genetic Engineering
and Biotechnology, Macedonian Academy of Sciences and Arts, Bul. Krste Misirkov 2,
POB 428, Skopje 1000, Republic of Macedonia; Tel.: +389-2-3235-411; Fax: +389-2-3115-434;E-mail: gde@manu.edu.mk
page: 55
|
|
RESULTS AND DISCUSSION
The molecular defects we detected in 35 of 50 (70%) patients, are shown in Table 1. Inversion in Intron 22. The inversion in intron 22 was identified in 19 patients, all with a severe form of the disease, giving an incidence of 38% of all screened patients, or 47.5% (19/40) of those with a severe form (Figure 1). Of these, 12 patients had a distal inversion, or type 1 (63.2%), and 7 (36.8%) had a proximal type of inversion, type 2. This finding agrees with the global frequency of this defect (23- 35% of all patients studied, and 41-57% of patients with a severe form of the disease) [12]. The mothers of all patients with this inversion were found to be heterozygous for the inversion. This accords well with the observation that it develops predominantly during male germ cell meiosis [13]. The intron 1 inversion was not detected in any of our patients. After the identification of the intron 1 inversion several years ago [5], a prevalence of 0-5% was reported [14]. This mutation was not found in 104 unrelated severe Hemophilia A patients from Hungary [15]. Alu Insertion. In one patient with severe disease, we detected an abnormal pattern using the cDNA probe covering exons 14-26 of the FVIII gene. Instead of a signal from a 5.9 kb fragment corresponding to exon 14, two prominent bands of ~3.6 kb and 2.6 kb, respectively, were present on the autoradiogram. This is indicative of an insertion of ~300 bp carrying a novel TaqI site within exon 14. Long-range PCR of the entire exon 14 of the patient, his brother, mother, maternal grandmother and a normal control, confirmed the presence of this inserted sequence of ~300 bp, which disrupts the reading frame at methionine 1224 in exon 14, leading to a stop codon within the inserted sequence. Sequence determination demonstrated that the insertion was a full Alu repeat belonging to the Yb8 family, the youngest family of Alu repetitive se quences [16]. Nucleotide Substitutions. All the other molecular defects we detected were single base substitutions (Table 2). Severe hemophilia was associated with five mutations predicted to cause premature termination or to affect the active site of the molecule. The change at codon 5 (CGA>TGA, Arg→Stop), was found in two unrelated, severely affected patients. Haplotyping of the FVIII gene in these two patients revealed the same haplotype, indicating a common founder. Three missense mutations (Arg 372→His; Arg1689→Cys; and Arg2163→His) were associated with severe disease. The Arg372→His mutation in exon 8 and Arg1689→Cys in exon 14, destroys the throm bin cleavage site. An Arg2163→His mutation in exon 23 was found in one patient. This mutation has been reported in both severe and moderate Hemophilia A patients (http:// europium. csc.mrc.58 ac.uk), indicating that it interferes with the molecular stability of the FVIII gene. All moderate cases were associated with missense mutations in different parts of the coding region (Table 2). The most abundant was an Arg531→Cys in exon 11, which was found in six (12%) unrelated patients. All mothers of these patients were carriers for the mutation, and all six patients shared the same haplotype (BclI+/Y; XbaI+/Y, intron 13 polymorphism 25/Y and intron 22 - 20/Y) indicating a founder effect. The other mutations (Asn90→Thr,Glu456→Val, Tyr473→His and Arg2159→ Cys) were found in individual families. Two new mutations of residues highly conserved in murine, porcine and canine FVIII were characterized. An AAC>ACC change at codon 90, exon 3, or Asn90→Thr, mutation was found in a patient and his cousin, with moderate disease (FVIII 3%). The mothers of both were carriers of the mutation, while their maternal grandmother had a normal DNA pattern (Figure 2). The maternal grandfather was not available for study but is clinically normal, thus suggesting a de novo FVIII mutation in his germ cells. A GAA>GTA change in exon 9, or Glu456→Val substitution, was found in a patient with moderate disease (Figure 3). Only one other GAA>AAA or Glu456→Lys at this position, has been described in a patient with moderate disease (http://europium.csc.mrc.ac.uk). The lower frequency of the known mutations is most likely due to the low sensitivity of the screening method used, especially SSCP. Also mutations could reside in regions that were not tested in this study (exon 14, promoter region or the 3 untranslated region) This is the first comprehensive study of the FVIII gene defects causing Hemophilia A in patients from Southeast Bulgaria. Based upon these results, it appears that carrier detection and prenatal diagnosis is possible in 70% of affected families. The two new mutations also increase our understanding of the molecular pathology of Hemophilia A.
Table 1. Molecular characterization of Hemophilia A patients from South East Bulgaria
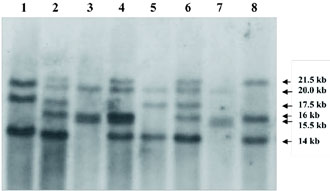
Figure 1. Southern blot analysis for detection of intron 22 inversion in the FVIII gene. Lanes 1 and 5: patients with inversion type 1; lanes 2 and 6: carriers for inversion type 1; lanes 3 and 7. patients with inversion type 2; lane 4: carrier for inversion type 2; lane 8: normal control.
Table 2. Mutations identified in the factor VIII gene among Hemophilia A patients from Southeast Bulgaria
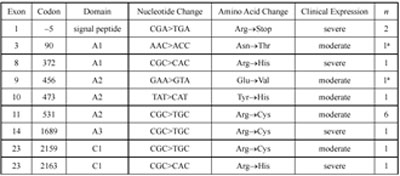
a These mutations are being reported for the first time.
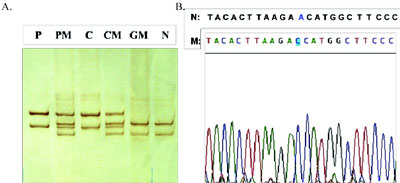
Figure 2. Representative photograph of SSCP analysis (A) and sequencing of abnormal fragment (B) identifying the new mutation AAC>ACC or Asn90.Thr at codon 90 of exon 3. Lanes P and C: patient and his cousin; lanes PM and CM: mothers of patient and cousin; lane GM: maternal grandmother; lane N: normal control.
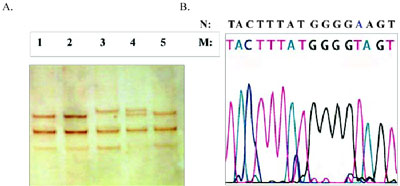
Figure 3. Representative photograph of SSCP analysis (A) and sequencing of abnormal fragment (B) identifying the new mutation GAA>GTA or Glu456.Val at codon 456, exon 9. Lane 3: patient; lane 4: his mother; lanes 1, 2 and 5: normal controls.
|
|
|
|

 |
Number 28
Vol 28 2025 Supplement |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|
|