

CHROMOSOMAL ABNORMALITIES IN
ENDOMETRIAL AND OVARIAN CARCINOMAS
PazarbaÕi A1,*, Kasap M1, Demirhan O1, Vardar MA2,
Suleymanova-Karahan D1, Doran F3
*Corresponding Author: *Corresponding Author: Ayfer PazarbaÕi, Ph.D., Department of Medical Biology and
Genetics, University of Çukurova, School of Medicine, 01330 Balcali, Adana, Turkey; Tel.:
+90-322-338-70-68; Fax: +90-322-338-65-72; E-mail: payfer@cu.edu.tr
page: 61
|
|
DISCUSSION
Understanding the molecular basis of the initiation and progression of ovarian and endometrial cancer will be strongly dependent on advanced cytogenetic and molecular genetics. In this cytogenetic study we described the chromosomal findings detected in 23 endometrial and ovarian tumors, only two had normal karyotypes, and possibly fibroblast-derived instead of tumorigenic tissues [12-14]. Although five tumors revealed only one simple structural change, the karyotypes of the other 16 tumors were quite complex. Our results indicate that several chromosomal regions were non randomly involved in abnormalities. The patients did not all show the same stage of histological grade and there was no relationship between the complexity of chromosome constitution and cancer progression.
Table 1. Chromosomal abnormalities in 12 endometrial (E1-12) and 11 ovarian (O1-11) carcinomas. The complete karyotypes are listed. When more than one karyotype was identified within individual samples, the number of cells of each type is shown in square brackets.
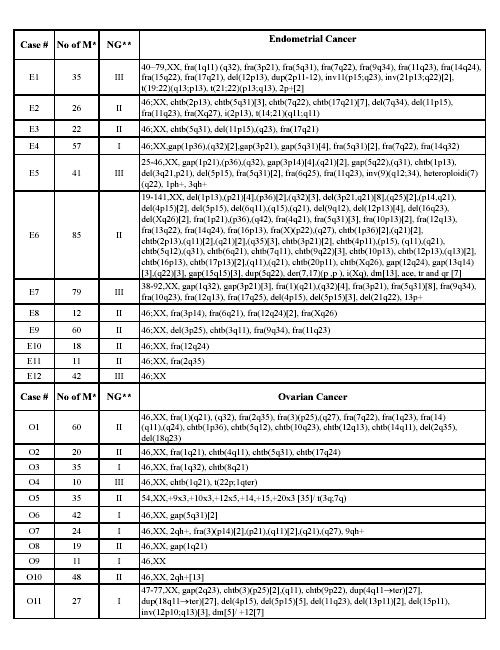
Table 2. Breakpoints and clonal alterations observed per chromosome in endometrial and ovarian cancer.Boldface represents cancer breakpoints.
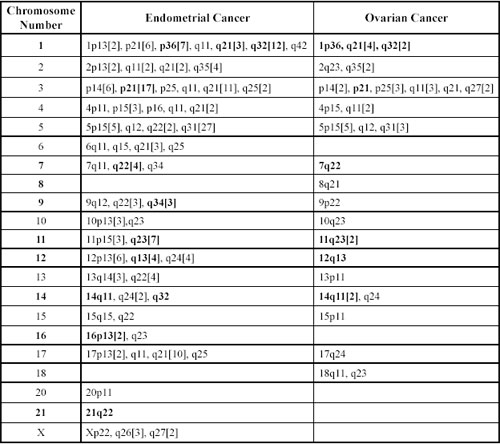
Chromosomes 3, 5 and 1 were (sequenced according to their frequency) most frequently involved in structural abnormalities in both endometrial and ovarian cases. Involved in structural clonal alterations were chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 10,11, 12, 13, 16, 17, 21, X in endo metrial cases, and chromosomes 1, 3, 5, 12, 14 in ovarian cases. In endometrial cases, deletion of chromosome 3, the most frequent structural alteration observed, was found in 13 cells of cases E5, E6 and E9, while no deletion of chromosome 3 was found in ovarian cancers. Deletions generally cause the loss of tumor suppressor genes [15-19]. Molecular studies have shown loss of heterozygosity (LOH) on 3p, which seems to be a common event in endo metrial tumors and related to LOH in the FHIT (fragile histidine triad) and the hMLH1 locus [13]. According to Imamura et al. [13], these losses contribute to the progression of the tumor to an undifferentiated stage, aggressive behavior, or metastatic potential. In another study [14], various deletions (3p13, 21, 22, 26) of chromosome 3p were associated with the development of uterine cervical carcinoma in Indian patients. Del(3)(q13q23) was the most consistent aberration observed in six of nine gastric and esophageal adenocarcinomas [15]. Through positional cloning of the 3q21 breakpoint, a novel gene, DIRC2, reported to encode a multi membrane-spanning protein that represents a new member of the major facilitator superfamily (MFS) of transporters, and these fragilities associated with sporadic and familial renal cell carcinomas [16].
Although the second most frequent clonal alterations that we observed was located on chromosome 5, the most frequent clonal alteration we observed in both tumor groups was 5q31. The q31 band of chromosome 5 has been the subject of intensive studies as it is a gene-rich area. It contains genes of known biological function, including growth factor, growth factor receptor, hormone receptor and neurotransmitter receptor [17].
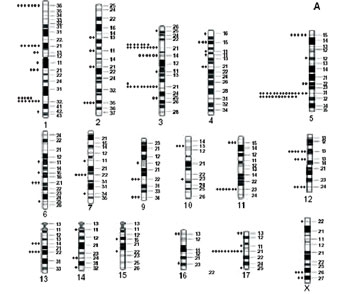

Figure 1. Localization of breakpoints and clonal alterations observed in endometrial (A) and ovarian (B) cancers.
Table 3. Frequency of abnormalities observed per chromosome in endometrial cancer (listed in decreasing order).
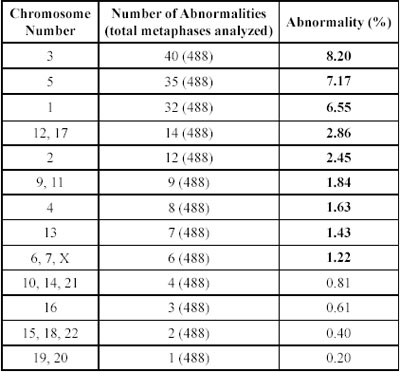
Table 4. Frequency of abnormalities observed per chromosome in ovarian cancer (listed in decreasing order).
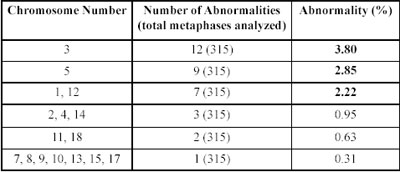
The third most frequent clonal alteration that we observed was located on chromosome 1. One of the clonal alteration in endometrial cases was at 1p36. Four genes, mapping to 1p36, have been proposed as candidate tumor suppressor genes: Heir-1 (ID3), a neuroectodermally expressed member of the Id family of developmental negative regulatory genes; TNFR2, one of two tumor necrosis factor receptor genes; PITSLRE, a cell cycle-regulated kinase gene with homology to human CDC2; and DAN, a transcription factor gene homologous to a mouse tumor suppressor gene. Two additional genes, the transcription factor regulator E2F2 and the paired-box-containing gene PAX7, were recently mapped to 1p36 and are thus additional potential candidate suppressor genes. Disruption of one or more of these candidate genes could play a role in the development of endometrial cancer [6].
The 12q13-15 region was involved in six cells of three cases in our study. It contains several genes (including an oncogene) of potential importance for neoplastic development, for example, SAS, SP1, INT-1, GLI, CHOP, CDK4 and MDM2 [19-21]. Molecular studies have shown that these genes are often coamplified, although there have also been cases with only one or two of these genes included in the amplicon [20,21]. The presence of an additional copy of chromosome 12 with an altered gene at 12q13 may contribute to the development of a malignancy. The HMGI-C gene encodes a member of the high-mobility-group proteins, which are non histone nuclear proteins able to bind AT-rich regions in the minor groove of DNA through three so-called AT-hook domains and may affect transcription by acting as architectural proteins. They have been found overexpressed in human and experimental malignancies [21]. Rearrangements of the HMGI-C gene are the consequence of chromosomal aberrations involving the 12q13-15 region where the HMGI-C gene is located [21].
Based on data on breakpoints at the 2q35-q37 region, a genetic link between some carcinomas has been suggested for a common pathway of the development of these tumors. This region include paired box protein (PAX3) gene [22]. However, the role of the PAX3 gene in rearrangements in chromosome 2 in endometrial and ovarian carcinomas remains to be determined.
The THY1 gene was mapped to the 11q23~24, which has been identified by both Taetle et al. [1] and Garba et al. [22] to have a high incidence of LOH in ovarian cancer. The location of the THY1 gene at this region makes this a suitable tumor suppressor gene for ovarian cancer. The THY1 gene was found to be exclusively expressed in the two non tumorigenic cell clones [23]. In contrast, THY1 expression was not determined in the tumorigenic ovarian cell lines. We observed clonal alterations at this region in both carcinoma types in our study. The mechanism of how THY1 expression is abolished in ovarian cancer cells remains elusive.
We observed fragility at 7q22 in both endometrial and ovarian tumors. That observed in endometrial tumors was a clonal alteration. Genes mapped to the 7q22 region include the human cut-like1 gene (CUTL1), human mis match repair gene (PMS2L), erythropoietin gene (EPO), and asparagine synthetase gene (ASNS). The fact that most structural rearrangements involving 7q22, suggest the hypothesis that this site contains a critical region of single or multiple genes, that when mutated or deleted results in malignant transformation. Whether this involves a structural alteration of a putative tumor suppressor gene remains to be clarified [24].
Two main cytogenetic subgroups have been identified in published cases of ovarian tumors with chromosome abnormalities: tumors with simple karyotypic changes and tumors with complex karyotypic abnormalities. Simple karyotypic changes are defined as simple numerical changes, whereas complex karyotypic changes are multiple numerical and structural abnormalities of chromo somes. We found that aberrations may be of significance in the pathogenesis of ovarian cancer and may provide valuable information for further investigation at the molecular level. Simple karyotypic changes, including trisomy 12, 10, 7, 8, 5 (in decreasing order of frequency) have been considered as primary chromosomal changes in ovarian cancer [5]. The 54,XX karyotype with complex numerical changes at chromosomes 9, 10, 12, 14, 15 and 20, which we found in case O5, have not been previously reported. These chromosomes may play a specific role in the patho genesis of ovarian cancer. Because trisomy 10 has been observed in some benign tumors and in normal tissues that surround malignant tumors, it may contribute to the trans formation of normal tissues and/or the initiation of tumori genesis [5]. Marked aneuploidy represents this aggressive phenotypic behavior at a cytogenetic level. However, chromosomal aberrations need not necessarily be associated with advanced-stage disease. We observed two normal 46,XX karyotypes in a highly malignant endometrial (E12) and an ovarian (O9) cancer.
In conclusion, this study has shown that specific chromosome bands, frequently involved in clonal alterations and breakpoints, may indicate the regions in which to search for new dominant oncogenes or recessive tumor suppressor genes. Our cytogenetic findings provide strong evidence that multiple genetic lesions are associated with the development of endometrial cancers, and that deletions of 3p13, p21, p23, q21 and q25 may play a specific role in the pathogenesis of such cancers. Chromosomal abnormalities have been known to occur in malignant tumors for a long time [1-5]. This study adds little to the understanding of the field of ovarian cancer cytogenetics; the results for endometrial cancer are more novel and could be improved by analyses of more cases.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|