

THALASSEMIAS AND OTHER HEMOGLOBINOPATHIES
IN FORMER YUGOSLAVIA
Efremov GD
*Corresponding Author: Professor Dr. Georgi D. Efremov, Research Centre for Genetic Engineering
and Biotechnology, Macedonian Academy of Sciences and Arts, Bul. Krste Misirkov 2,
POB 428, Skopje 1000, Republic of Macedonia; Tel.: +389-2-3235-411; Fax: +389-2-3115-434;
E-mail: gde@manu.edu.mk, gdefremov@yahoo.com
page: 11
|
|
RESULTS AND DISCUSSION
Population Surveys. Before the National Reference Laboratory for Hemoglobinopathies (NRLH) was established in 1970, the frequency and distribution of various thalassemia types and other hemoglobinopathies in the Republic of Macedonia were determined by the Department of Pediatrics, Faculty of Medicine at Skopje. From 1965 to 1967, blood samples from 2,861 apparently healthy school children, aged 8 to 18 years, from 32 different parts of the country, and 561 cord blood samples, obtained through the services of the Department of Obstetrics, Faculty of Medicine, Skopje, Republic of Macedonia, were studied [15]. The gene for β-thal was found to be distributed through most of the country, with a frequency from 1 to 20% (mean value 4.7%). We also detected six cases (0.21%) with three different abnormal Hbs that were later characterized as Hb J-Paris-I [α12(A10)Ala→Asp, GCC>GAC (α1 and α2)], Hb Beograd [β121(GH4) Glu→ Val, GAA>GTA] and the Lepore Hbs [16,17]. After 1970, we screened 2,201 school children from the region of Gevgelija and Bitola [18] and 2,853 children from Strumica, Radovis and Stip [19]. The incidence of β-thal and other hemoglobinopathies in these regions was 9.5, 2.4, 6.0, 5.3 and 3.5%, respectively. Between 1980 and 1982, we screened 2,498 school children of both sexes living in Skopje [20]. Skopje was selected because its population had increased four-fold in the previous 30 years. In fact, one quarter of the Macedonian population live in this city. Eighty-one children were carriers of β- and δβ-thal trait, Swiss type of hereditary persistence of fetal Hb (HPFH), and Hb S [β6(A3) Glu→Val, GAG> GTG], Hb O-Arab [β121(GH4) Glu→Lys, GAA>AAA], Hb D-Punjab [β121(GH4) Glu→Gln, GAA>CAA], Hb Strumica [α112(G19) His→Arg (CAC>CGC) (α2 or α1)] and Hb Lepore- Washington-Boston (Lepore-WB; δ87Gln- β116His). The incidence of β-thal was 2.4%, and that of the observed types of hemoglobinopathies was 3.2%. This incidence seemed to be the most likely one for the Macedonian population [17,20]. Between 1992 and 1994, 3,482 school children from the northern and west-northern parts of Macedonia were screened. Heterozygous β-thal was detected in 30 children (0.9%), δβ-thal in eight (0.2%), α-thal in 26 (0.7%), Swiss type of HPFH in eight (0.2%), and four carriers of abnormal Hbs (0.1%). The overall incidence of all hemoglobin opathies was 2.2%. The incidence of α-thal and the organization of the γ-globin genes was studied in cord blood samples from 3,232 newborns [21], received from the Department of Obstetrics and Gynecology, Faculty of Medicine, Skopje, and Hospital for Obstetrics and Gynecology Cair, Skopje, Republic of Macedonia. Twenty-six newborns were found to be carriers of α-thal (0.8%), of whom 25 were carriers of α-thal-2 and one of α-thal-1. In addition, eight babies were carriers of an abnormal Hb, three with Hb O-Arab, and one each with Hb Hamilton [β11(A8) Val→Ile, GTT>ATT], Hb Strumica and the newly de scribed γ chain variants: Hb F-Macedonia-I [Aγ2(NA2) His→Gln, CAT>CAG] and Hb F-Macedonia-II [Gγ104 (G6)Lys→Asn, AAG>AAC] [22,23]. These five population surveys on a total number of 22,136 subjects, gave an overall incidence of β-thal and other hemoglobinopathies in the Republic of Macedonia of 3.4%, varying between 0.7% and 12.3%. Screening of 2,700 individuals from Serbia, 1,200 from Vojvodina, 1,611 school children from Kosovo, 7,601 individuals (most of whom exhibited signs of hemo lysis and/or low RBC indices) from Croatia, 2,780 school children from Bosnia and Hercegovina, and 860 school children from Monte Negro, gave an incidence of β-thal and other hemoglobinopathies in these territories of the Former Yugoslavia of 2.3, 2.0, 2,7, 4.4, 2.7, and 2.6%, respectively [24]. The frequency of β-thal in Former Yugoslavia varies from 1.2% in the north (Vojvodina) and 2.9% in the south (Macedonia) [24]. The frequency of β-thal in Serbia is 1.8%, in Bosnia and Hercegovina 1.5% [24,25], 1.9% in Monte Negro, and 2.0% in Kosovo [24]. Figure 1 shows a map of the Former Yugoslavia with the number of subjects examined and frequency of β-thal trait, and combined frequency of β-thal trait and other hemo globinopathies, in the different republics and provinces. 14 Hematological and Biochemical Characteristics of β-Thalassemia. The hematological and biochemical changes seen in the β-thal heterozygotes vary: some carriers are practically free of complaints with or without a slight anemia, while others have a moderate anemia. Microcytosis and hypochromia with low MCV and MCH values are, however, always present, and the resistance of the erythrocyte to saline solutions is increased. Figure 2 illustrates the MCV and MCH values in β-thal heterozygotes and normal controls studied in our laboratory. The MCV value, independent of the type of mutation, averaged 68.2 fL in comparison to 92.9 fL in normal individuals. The average MCH value was 21.0 pg in β-thal heterozygotes and 30.9 pg in normal individuals. Relatively large variations in the MCV and MCH values exist (56.0-77.0 fL and 17.0-27.0 pg, respectively) which is likely to be due to individual variations and to the type of mutation. Table 1 gives the results of some of our hematological and Hb analysis data for β-thal heterozygotes with 15 different mutations. The resistance to saline solutions of the erythrocytes from heterozygotes for β-thal was increased. Our results on a one-tube osmotic fragility method that used three buffered solutions (0.32% saline, 0.36% saline and Tyrode solution) as a screening test for β-thal trait, published in 1981 [8], showed that 0.36% saline was the most sensitive and most effective percentage as it detected 96-100% of heterozygotes with β-thal compared to about 80% with both 0.32% and Tyrode solution. Analysis of the in vitro Hb synthesis in the reticulo cytes of β-thal homozygotes and heterozygotes was very useful in estimating the globin chain imbalance due to decreased or absent β chain production. Table 2 summarizes our results for the globin chain synthesis of 47 β-thal patients homozygous for 15 different mutations. In β-thal heterozygotes, the synthesis of the β chains was about half that of the α chains. The degree of the β chain deficiency was related to the type of mutation, and it varied from 0.44 in heterozygotes for the codon 39 (C>T) mutation to 0.65 in heterozygotes for the nt 30 (C>T) mutation (see Table 1). Forms of Thalassemia and Related Conditions. The biochemical expressions of the homozygous and double heterozygous states of β-thal and related conditions were studied simultaneously with the population surveys. The homozygous state of β-thal was first recognized in the Republic of Macedonia in 1952 [26]. From then until 1974, 95 patients with Cooleys Anemia were registered at the Department of Pediatrics, Faculty of Medicine, Skopje, Republic of Macedonia [27]. A patient with Hb H disease [28], nine families with Hb Lepore [29], and two families with different expressions of homozygous β-thal [30], were also described. As of 2008, 157 cases of thalassemias major have been registered at the Department of Pediatrics, Faculty of Medicine, Skopje, Republic of Macedonia. Since 1970, when the NRLH was established, studies of patients with hemolytic anemia and cyanosis included patients from all the territories of Former Yugoslavia. Table 3 summarizes the types of thalassemia and related conditions detected and characterized through studies of patients with signs of hemolysis and/or low RBC indices. Two-thirds of the patients were from the Republic of Macedonia and Croatia, while one-third were from the other territories of Former Yugoslavia. Twenty-three types of thalassemia and related conditions were detected. The most common type was heterozygous β-thal, observed in 1,044 patients (74.1%) of all the cases with hemoglobin opathies. The 11 different types of thalassemia major and the five different types of thalassemia intermedia were characterized in 132 and 15 patients, respectively. Heterozygous δβ-thal was detected in 162 individuals, Hb Lepore in 149 individuals, and the Swiss type of HPFH in 131 persons. Our results clearly indicate that hemoglobinopathies are heterogeneously distributed in the Former Yugoslavia. The frequency of β-thal and related conditions ranges from 0.1% in the northwest (Slovenia) to 3.4% in the south (Republic of Macedonia). Although the national boundaries of the republics of Former Yugoslavia do not necessarily represent particular ethnic groups, our results indicate that hemoglobinopathies are rare in Slovenians, common in Croatians and Serbs, and very common in Macedonians. Molecular Characterization of β-Thalassemia and Related Conditions. The molecular basis of β-thal in Former Yugoslavia was studied in great detail. Table 4 lists the 27 different β-thal alleles that were discovered in a total of 705 β-thal chromosomes, of which 508 were from Macedonian, 46 from Serb, 53 from Croatian, 62 from Albanian, and 36 from Turkish patients. Sixteen β-thal alleles were identified in Macedonian patients, and 8, 14, 8 and 11 alleles, respectively, were identified in the Serbian, Croatian, Albanian, and Turkish patients. In each of the ethnic groups, 5 to 7 different alleles account for over 85% of all the characterized alleles. The most common alleles are IVS-I-110 (G>A), IVS-I-6 (T>C), IVS-I-1 (G>A) and codon 39 (C>T) in the Macedonian, Serb and Croatian patients. The most common alleles in the Albanian patients were codon 39, nt 30 (T>A), IVS-I-6 and IVS-I-110, and in the Turkish patients, codons 8/9 (+G), codon 8 (AA), codon 39, IVS-I-110 and IVS-I-6. The number of the different β-thal alleles was much smaller in the Serbian and Albanian patients. Nine of the 27 β-thal mutations, were discovered for the first time: nt 101 (C>T) in a Bulgarian patient [13], nt 30 (T>A) in a Macedonian β+-thal/ Hb Lepore patient [31], G>A at position +22 3 to the Cap site in Bulgarian and Turkish patients [32], initiation codon mutation ATG>ACG in a Macedonian heterozygote [33], codon 6 (A) in a Bulgarian thalassemia major patient [14], codons 82/83 (G) in a β-thal heterozygote from Croatia [34], IVS-II-850 (G>C) in a Croatian family heterozygous for β-thal [35], polyadenylation signal (poly A) (AATAAA>AATGAA) in a Bulgarian patient with thalassemia major and one Macedonian patient with thalassemia intermedia [36], and a 1.6 kb deletion of the β-globin gene in a Croatian β-thal heteozygote [37]. Molecular characterization of the mutation in patients with thalassemia major, intermedia and minor, resulted in the identification of deletional types of β-thal. In 1986, we described a new type of deletional δβ-thal, named the Macedonian δβ-thal [38], a new type of εγδβ-thal, named Croatian εγδβ-thal [12], a deletional type of β-thal, named Croatian β0- thal [37], and a new type of γ-globin gene triplication [11]. δβ-Thalassemia. δβ-Thal results from simultaneous deficiency in synthesis of δ- and β-globin chains from an anomaly in the closely linked δ- and β-globin genes. It is characterized by normal levels of Hb A2 and increased levels of Hb F that is unevenly distributed in the red cells. The hematological and clinical findings in δβ-thal heterozygotes are similar to those observed in classical β-thal heterozygotes. Although classical β-thal is present in moderately high frequencies in the Macedonian population, no cases of δβ-thal were reported until 1973. In a survey of 5,500 school children from southern parts of the Republic of Macedonia, only one hetrozygote for δβ-thal was detected [39]. In 1975, we described the first two Macedonian families with δβ-thal, in which two members were heterozygous for both β- and δβ-thal [40], and soon thereafter, we described a Serbian family with δβ-thal interacting with Hb Lepore-Baltimore (δ68→β84) [41]. The results of hematological and biochemical analyses of two patients homozygous for δβ-thal and 219 patients heterozygous for δβ-thal were presented at a meeting [42] but never published. The two δβ-thal homozygotes were members of Croatian families originating from Rijeka and Sisak, Croatia. The first was a 30-year-old woman, and the second patient was a worker, age 28. Both had hemolytic anemia and hepato splenomegaly. The hematological analysis for the woman gave the following results: RBC 4.48 × 1012/L, Hb 11.5 g/dL, PCV 0.346 L/L, MCV 77 fL, MCH 26 pg, moderate hypochromia, poikilo- and anisocytosis. Starch gel, cellulose acetate and agar gel electrophoreses showed the presence of Hb F only. In vitro biosynthetic experiments showed the presence of γ- and α chain only. The relative radioactivity of γ to α chain was 1.91, and the specific activity 1.67. The results of hematological and Hb analyses of over 300 heterozygotes for δβ-thal are presented in Table 5. The Lepore Hemoglobinopathies. This was the most common hemoglobinopathy in Former Yugoslavia. The first nine unrelated families with Hb Lepore were de scribed in 1968 [29], both in homozygous and double heterozygous states, interacting with β-thal states, and characterized as Hb Lepore- Washington [16,17,24]. Hematological and biochemical analysis of blood samples from a patient at the Department of Internal Medicine, Faculty of Medicine, Belgrade, Serbia, and her parents and relatives, disclosed the presence of Hb Lepore-Baltimore interacting with δβ-thal [41]. During the past 40 years, we have studied 163 unrelated patients or families with Hb Lepore-Washington-Boston, i.e., nine homozygotes, 17 compound heterozygotes, and 137 heterozygotes. The symptoms and blood films in heterozygotes and homozygotes do not differ greatly from those found in equivalent β-thal states. The homozygous state for Hb Lepore was observed in six Macedonian, two Serb and one Croatian patients. All suffered from severe hemolytic anemia; eight died between the ages of 5 and 14 years, and one at 24 years. The clinical expression of the compound heterozygotes for Hb Lepore and β-thal varies from very severe to moderate anemia, and is determined by the type of the β-thal mutation [43]. The clinical findings for the homozygotes are given in Table 6, and their hematological and Hb data in Table 7. In the heterozygotes, about 10% of the Hb is Hb Lepore, the level of Hb A2 is normal or slightly decreased, and that of Hb F is slightly increased in about two-thirds of subjects. Hereditary Persistence of Fetal Hemoglobin. Hereditary persistence of fetal Hb was first reported in Former Yugoslavia in 1969 [15]. Of the 2,861 subjects screened, 35 (1.2%) had elevated levels of Hb F (mean 3.7%, range 1.5-8.6%) and a normal level of Hb A2, normal RBC indices and normal osmotic fragility. A second type of HPFH was characterized by a high Gγ level in the Hb F and by triplication of γ-globin genes (Gγ.Gγ.Aγ/Gγ.Aγ) [12]. Many other cases appeared to have the Swiss type of HPFH (1.1%), which seems to be present mostly in Macedonians [23]. DNA haplotyping of the β-globin gene cluster from 23 unrelated families with the Swiss type of HPFH showed it to be associated with a haplotype identical to that of the Senegal (#3 [ + + + + + + +]) type [12]. α-Thalassemia. The first survey for α-thal, undertaken on 561 cord blood samples, showed an incidence of 3.8%. Studies of a further 1,140 newborn babies per formed in 1980-81, showed an incidence of α-thal-1 of 0.8%, and for α-thal-2 of 2.4% [44]. A recent survey of 3,326 cord blood samples from Macedonian, 1,546 Croa tian, 1,043 Albanian, 373 Gypsy and 99 Turkish new borns, gave incidences for α-thal-1 of 0.42, 0.39, 0.10, 0.0, 0.0 and 0.30, respectively, and for α-thal-2 of 1.89, 1.03, 0.10, 0.0, 0.0 and 1.24, respectively (unpublished data). Molecular characterization resulted in the identification of four different types of α-thal: α3.7/, αMED Iα/ (17.5 kb), α20..5/ and Hb Icaria [α142, Term→Ser (TAA>TCA in α2)] [45-47]. Abnormal Hemoglobins. Table 8 lists the Hb variants detected in Former Yugoslavia. Twentyeight different variants, in 253 unrelated individuals or families, were characterized. Of these, four were α chains, 18 β chains, two γ chains, one δ chain, one extended chain and two fusion chain variants. Five were unstable and two were hyperunstable variants causing a severe hemolytic anemia, two were metHb variants causing methemoglobinemia, and two were fusion chain variants causing anemia similar to that found in the thalassemias. As already mentioned, the most common variant is Hb Lepore (Lepore-WB) that is distributed throughout Former Yugoslavia and has been observed in 163 unrelated individuals or families. Hb O-Arab is the second most common variant, being found mainly in the Republic of Macedonia, where it has been observed in 22 Gypsy and six Albanian families. This variant has been observed in three instances in the homozygous state [24,48], and in two instances in association with β-thal [24]. Hb Beograd [49,50], and Hb Strumica [51], seem to occur only in Yugoslavia, and have also been found in Yugoslavian families living in Australia and New Zealand [52], and in Turkish families who emigrated from Yugoslavia [53]. Seven variants, i.e., Hb Leiden [β6(A3) or β7(A4) Glu→0], Hb Yokohama [β31(B13)Leu→Pro, CTG>CCG], Hb Sabine [β91(F7)Leu→Pro, CTG>CCG], Hb Koln [β98(FG5)Val→Met, GTG>ATG], Hb Beth Israel [β102(G4)Asn→Ser (AAC>AGC)] and Hb Bush wick [β74(E18)Gly→Val, GGC>GTC], that were found in Former Yugoslavia, have arisen as de novo mutations.
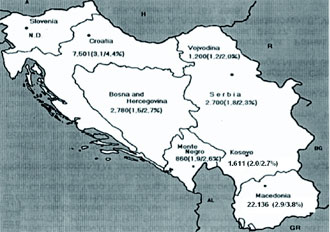
Figure 1. Map of former Yugoslavia showing the number of screened school children and adults (38,888) and frequency of β-thal and β-thal and other hemoglobinophaties (in parentheses). Most of the subjects from Croatia were patients with signs of hemolysis and/or low RBC indices. N.D. = not determined
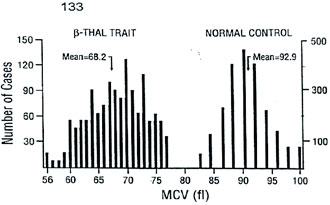
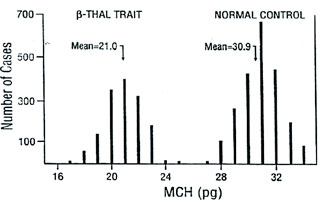
Figure 2. Distribution of MCV and MCH values for 1,500 â-thal heterozygotes and 2,175 normal individuals.
Table 1 Hematological and hemoglobin composition data for â-thalassemia heterozygotes with 14 different mutations (average values only)
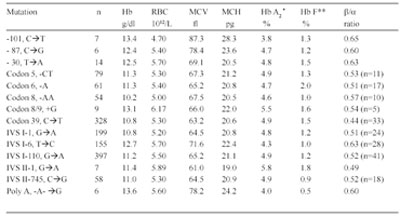
* Determined by microcolumn chromatography; ** Determined by alkali denaturation
Table 2 In vitro Chain Synthesis Data for â-Thalassemia Homozygotes and Compound Heterozygotes with Different Mutations*
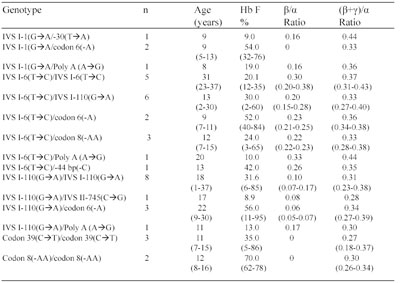
* Most of the patients were on a regular blood transfusion regimen. Analyses were performed at least 30 days after the last transfusion.Values between parenthesis identify ranges or normal values.
Table 3 Forms of Thalassemia and Related Conditions Detected in Former Yugoslavia (December 2007)
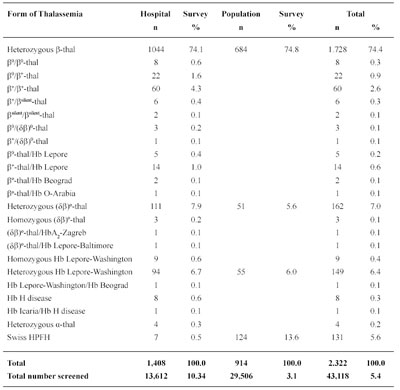
Table 4 Frequencies of the different â-thalassemia alleles among Macedonian, Serbian,Croatian, Albanian, Turks identified in NRLH-RCGEB-MASA
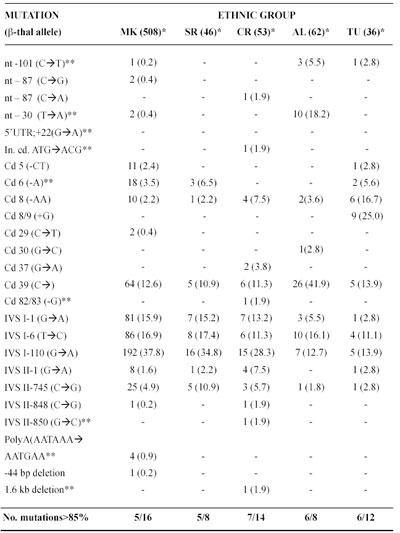
MK=Macedonian; SR=Serbian; CR=Croatian; AL=Albanian living in Macedonia and Kosovo; TU=Turks living in Macedonia.*No. of chromosomes studied; **New mutation identified in the RCGEB MASA, Skopje, R. Macedonia
Table 5 . Hematological and Hb analysis data for heterozygotes for δβ-thalassemia
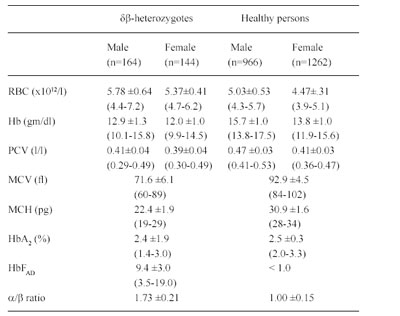
Table 6 Clinical findings in nine homozygotes for Hb Lepore
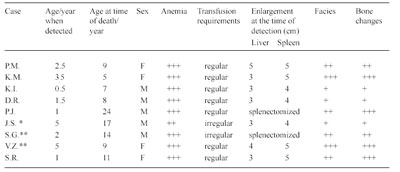
+ = mild; ++ = moderate; +++ = severe; *patient from Zagreb; ** patient from Beograd
Table 7 Hematological and hemoglobin composition data in nine homozygotes for Hb Lepore.
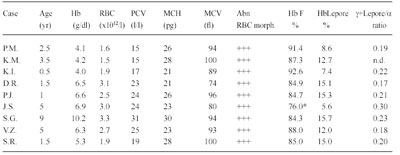
* Tested one month after last transfusion
Table 8 Abnormal Hemoglobind Detected in Former Yugoslavia
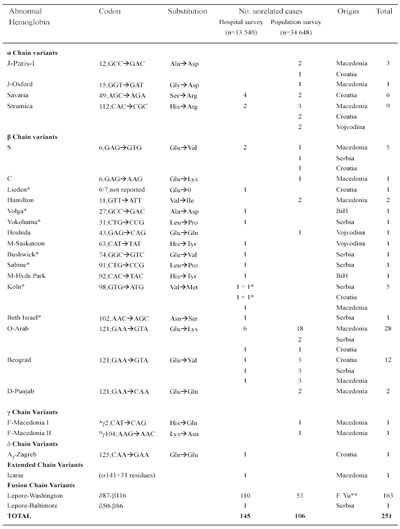
* De novo mutation; ** In all republics and provinces of former Yugoslavia
|
|
|
|

 |
Number 28
Vol 28 2025 Supplement |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|
|