

MOLECULAR ANALYSIS OF A FAMILY WITH
CONGENITAL ADRENAL HYPERPLASIA
GENOTYPE/PHENOTYPE DISCREPANCY
Anastasovska V, Kocova M*
*Corresponding Author: *Corresponding Author: Professor Dr. Mirjana Kocova, University Pediatric Clinic, Medical Faculty,
Vodnjanska 17, 1000 Skopje, Republic of Macedonia; Tel.: +389-2-3111713; Fax: +389-2-3229-027;
E-mail: mirjanakocova@yahoo.com
page: 23
|
|
RESULTS AND DISCUSSION
To distinguish between the inactive pseudogene (CYP21P) and the active gene (CYP21), two pairs of oli gonucleotide primers (21AF/AR and 21BF/BR, respectively) were used for differential amplification of these genes as previously described [17]. A 3.2 kb product was detected (Figure 1).
The PCR products generated from CYP21P and CYP21 were distinguishable by digestion with the restriction enzyme EcoRI, which for the CYP21P produced three fragments (0.5, 0.6 and 2.2 kb), whereas for the active CYP21 gene produced only two fragments (1.0 and 2.2 kb). The primary PCR products of the CYP21 gene were subjected to locus analysis by using secondary ACRS PCR (Table 1).
Table 1. Amplification-created restriction site detection of mutations in the CYP21 genes
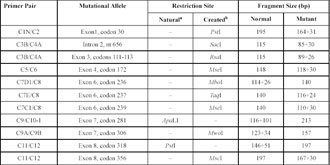
a Natural: in two cases, the mutations had naturally created recognition sites for direct restriction detection.
bCreated: region-specific primers for nine known mutation loci were designed that would lead to the creation of new restriction recognition sites at these known mutation sites on secondary amplification.
Two of the 11 CAH mutations analyzed were detected in the proband and in her parents (Figure 2). The proband was heterozygous for two mutations, one in exon 1 (at codon 30, which is considered to be a mild mutation), the second in exon 8 (at codon 318, considered to be a severe mutation). This genotype usually produces a moderate phenotype of the disease. The mother was homozygous for the mutation in exon 1 (at codon 30) and only had difficulty in conceiving. The father was homozygous for the mutation in exon 8 (at codon 318), a nonsense mutation usually associated with severe salt-wasting, and only suffered from difficulty in fathering a child, which is an unusual presentation of the disease. These results indicated that the proband inherited the mutation in exon 1 at codon 30 from her mother and that in exon 8 at codon 318 from her father.
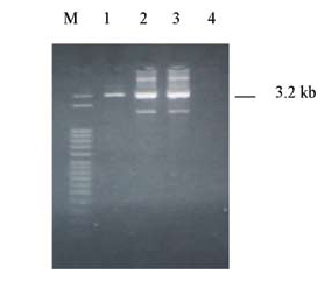
Figure 1. The CYP21 gene (3.2 kb). Lanes 1, 2, 3; lane 4: blank; M: marker (50 bp).
A clear genotype/phenotype correlation in CAH patients has been established by many large studies [18,19]. Classification of mutations into severe, moderate and mild, and their combination, can predict the severity of the clinical presentation in approximately 79% of patients [18]. However, there is a high degree of overlap between moderate and mild forms. The Pro30Leu anomaly, usually associated with a simple virilizing form of the disease, is one of the less frequent mutations, and has also been described in non classical mild forms of the disease [19]. Our patient carried one severe mutation (codon 318) and one moderate mutation (codon 30), and presented with a mild form of late onset and mild evolution. From her genotype, we would expect a more severe form of the disease with earlier presentation and more severe virilization. Treatment was effective in the progression of the puberty and bone maturation.
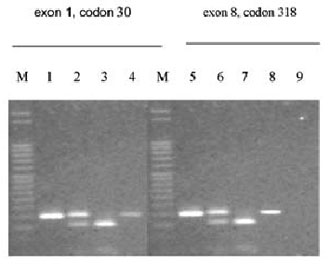
Figure 2. Mutations in exon1/codon 30 and exon 8/codon 318 of the CYP21 gene. M: marker (50 bp); lanes 1 and 5: PCR II product; lanes 2 and 6: patient; lanes 3 and 7:mother; lanes 4 and 8: father; lane 9: blank.
Both parents are also CAH patients on the basis of genotype. The mother, who was homozygous for the mild mutation (codon 30), did not have symptoms of the disease until the age of conception, when she was unable to conceive and was treated for sterility. No history of early virilization was present. Her clinical presentation is consistent with the genotype, although earlier presentation would have been expected.
The father, homozygous for the severe nonsense mutation (codon 318), which usually presents with a salt-wasting form accompanied by precocious puberty and fre quently by sterility. Although improvement of salt-wasting has been shown with age in some studies [19], we were not able to find another published case of homozygosity for the nonsense mutation who survived childhood without therapy. All inquiries about the fathers early puberty were also negative. Thus, his only presentation was a poor sperm count. De novo recombination and unequal crossover involving CYP21 have been documented in sperm [19], however, there is no evidence of this process in our patient. It is impossible to postulate which parent contributed most to the failure to conceive a child spontaneously.
One might speculate that there may be additional 21-hydroxylase activity not encoded by the CYP21 gene that contributes to the moderate clinical presentation. that contributes to the moderate clinical presentation. Modifying genes and transcription abnormalities of the CYP21 gene could also be involved.
Genotype/phenotype discrepancies pose difficulties, especially for genetic counseling of families. An extended pedigree with genotyping of more members of this family might be helpful in the future.
In conclusion, based on the genotype, a more severe phenotype would have been expected in the mother, the father and the child. It is possible that some protective mechanisms, yielding higher 21-hydroxylase enzyme activity, have been transmitted to the child from both parents. Further analyses of the enzyme activity are necessary to elucidate the unusual clinical presentation and genotype/phenotype discrepancies.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|