Neurodegenerative diseases are characterized by neu ronal loss and intraneuronal accumulations of fibrillary materials. Among several intracellular inclusions such as Hirano bodies, Lewy bodies, Pick bodies and neurofibril lary tangles (NFT), the latter are the most common [1]. They are consistently found in Alzheimers disease (AD), postencephalitic parkinsonism, amyotrophic lateral sclerosis/parkinsonism- dementia complex (ALS/PDC) of Guam, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Picks disease, frontotemporal dementia (FTD), dementia pugilistica and head trauma, and Downs syndrome. They have also been described in Gerstmann-Straussler-Scheinker syndrome (GSS), Hal-lervordern-Spatz disease, myotonic dystrophy (DM), Niemann-Pick type C (NPC) disease, subacute sclerosing panencephalitis among other rare conditions, but also occur in normal aging and contain the hyperphosphorylated microtubuleassociated protein tau [1]. In this review, the molecular bases of AD, postencephalitic parkinsonism,ALS/PDC, PSP, CBD, Picks disease, FTD, Downs syndrome, DM and NPC disease, all considered to be common tauopathies, will be discussed [1].
Alzheimers Disease (AD). Alzheimers disease is a progressive neurodegenerative disorder that leads to dementia and affects approximately 10% of the world population older than 65 years of age. Memory loss is followed by aphasia, agnosia, apraxia and behavioral disturbances [1]. Senile plaques and NFT are the main brain lesions observed in AD. The former result from extracellular accumulation of the peptide Aâ, derived from the â-amyloid precursor protein (APP), into amyloid deposits, which are diffusely and variably distributed throughout the cerebral cortex and in subcortical structures [2,3]. In familial AD, mutations have been found on the APP gene [2]. The NFT consist of abnormal fibrils aggregated into paired helical filaments (PHF) and contain the hyperphosphorylated form of tau protein. They are usually observed in the large pyramidal cells of the hippocampus and entorhinal cortex, the supragranular and infragranular layers of the association cortical areas, nucleus basalis of Meynert, amygdala, locus coeruleus and dorsal raphe, the primary motor and sensory cortices being relatively spared. The demonstration of both senile plaques and NFT within these regions of the cerebral cortex is essential for a definite diagnosis of AD. However, NFT at a lower density are also present in the entorhinal cortex and hippocampus of normal elderly brains, whereas the neocortex exhibits only isolated NFT [1,3].
Postencephalitic Parkinsonism. Many patients who survived the influenza pandemic of 1916-1926 later developed postencephalitic parkinsonism with extrapyramidal symptoms as the major clinical features. Affected patients did not show any cognitive changes and were usually neither aphasic nor apraxic[1]. Immunohistochemical analysis demonstratedthat NFT were present in variable density in the hippocampus and entorhinal cortex, in neocortical areas and in subcortical regions. Higher NFT densities were observed in the hippocampus (CA1 and subiculum), area 20 of the neocortex and putamen, indicating that some regions were preferentially affected by the degenerative process [1,4].
Amyotrophic Lateral Sclerosis/Parkinsonism Dementia Complex (ALS/PDC) of Guam. This chronic neurodegenerative disorder is highly prevalent in the native Chamorro population of Guam in the Western Pacific [2]. Clinically, it is indistinguishable from sporadic ALS and presents with fasciculations and signs of involvement of lower and upper motor neurons. Parkinsonism-dementia is characterized by an insidious progressive mental decline and extrapyramidal signs including bradykinesia, rigidity and, less often, tremors. Both aspects of the disease are frequently associated but may occur separately. The brain shows severe cortical atrophy and neuronal loss. There is widespread NFT formation, especially in the temporal and frontal isocortex, hippocampal formation and several sub cortical structures [2,5]. Immunohistochemical studies have revealed that pathological tau proteins are present in the NFT [1].
Progressive Supranuclear Palsy (PSP). This late-onset atypical parkinsonian disorder is characterized by supranuclear vertical gaze paralysis, moderate or severe postural instability with episodes of falling during the first year of onset of symptoms, and facial, nuchal and tron cular dystonia; dementia is common in the later stages [1,5,6]. Progressive supranuclear palsy is characterized by neuronal loss, gliosis and NFT formation. Subcortical localization of NFT in basal ganglia, brainstem and cerebellum, led to PSP being considered as a model of "subcortical dementias." However, degenerative lesions have been described in the perirhinal, inferior temporal and prefrontal cortex, the density of NFT varying among cases [6,7]. Further studies have demonstrated that the primary motor cortex is more severely=affected than the neocortical association areas when compared to AD [1,5,8]. Glial fibrillary tangles have been detected in PSP brains [5,8-11].
Corticobasal Degeneration (CBD). This rare, sporadic and slowly progressive neurodegenerative disorder is clinically characterized by cognitive disturbances, e.g., aphasia and apraxia, and extrapyramidal motor dysfunction, e.g., rigidity, limb dystonia, akinesia and action trem or [1,5]. Sometimes moderate dementia emerges late in the course of the disease. There is clinical and pathological overlap between PSP and CBD, so that distinction of these conditions on a neuropathological basis becomes very important. Neuropathological examination shows fronto parietal atrophy of the brain and glial and neuronal abnormalities [5,11]. The glial pathology is dominated by astro cytic plaques, and numerous tau-immunoreactive inclusions in the white matter. The presence of achromatic ballooned neurons has been shown in cortex, brainstem and subcortical structures, as well as neuritic changes and NFT. Immunochemical studies have been given great importance in distinction of PSP and CBD [1,5,11].
Picks Disease. This rare disorder is characterized by a distinctly progressive dementia. Early in the clinical course, there are signs of frontal disinhibition including mood disturbances and progressive language impoverishment leading to mutism [1]. Prominent frontotemporal lobar atrophy, gliosis, severe neuronal loss, ballooned neurons and the presence of neuronal inclusions, called Pick bodies, in both cortical and subcortical structures are characteristic of the disease [1,2]. Pick bodies contain tau protein and occur at higher density in the hippocampus than in the neocortex. In the former, Pick bodies are numerous in the granule cells of the dentate gyrus, in the CA1 field, the subiculum and the entorhinal cortex. In the latter, they are mainly found in layers II and VI of the anterior segment of temporal and frontal lobes [1,2,5].
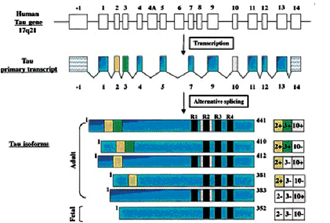
Frontotemporal Dementia (FTD). Historically, this has often been classified as a form of Picks disease, even when Pick bodies were not present. However, FTD may be categorized with different subgroups. A consensus on clinical and neuropathological criteria for FTD has been published which clari-fied the distinction between Picks disease and FTD [12]. An autosomal dominant disease related to FTD, characterized by adult-onset behavioral disturbances, frontal lobe dementia, parkinsonism and amyotrophy and linked to chromosome 17q21 has been described and is called FTD with parkinsonism linked to chromosome 17 (FTDP-17) [12]. Although clinical heterogeneity has been described between and within families with FTDP-17, the usual symptoms include behavioral changes, loss of frontal executive functions, language deficit and hyperorality [12,13]. Parkinsonism and amyo trophy have been found in a few families, but are not consistent features [14]. Brains of FTD patients show atrophy of frontal and temporal lobes, severe neuronal cell loss, grey and white matter gliosis, and superficial laminar spongiosis. An important characteristic is the filamentous pathology that affects neuronal cells and/or glial cells in some cases. Tau mutations segregate with the pathology of FTDP-17 indicating their pathogenic role [13-15].
Myotonic Dystrophy [DM]. This autosomal dominant and slowly progressive multisystemic disorder is characterized by myotonia, muscular atrophy, cataract and endocrine dysfunction. Affected individuals present with a highly variable phenotype, ranging from an asymptomatic condition to a severe congenital form. Impairment of intellectual and cognitive function in DM has been reported [16]. The molecular basis is an unstable CTG trinucleotide repeat in the 3 untranslated region (3UTR) of a gene that encodes a putative Ser/Thr protein kinase (DM protein kinase), located on chromosome 19 [16]. There is reduced brain weight and minor abnormalities in gyral architecture, and a disordered cellular arrangement with neurons being present in subcortical white matter, and intracytoplasmic inclusion bodies in cortical and subcortical structures. The presence of abnormally frequent NFT has been reported in the temporal lobe, especially in the parahippocampal gyrus [16]. Immunoblotting has revealed taupositive inclusion bodies in the hippocampus, the entorhinal cortex and in most of the temporal areas.The amounts of tau proteins are higher in the most severely affected brains, but lower than in AD [1].
Downs Syndrome. Downs syndrome patients have numerous somatic dysfunctions that occur during development which are due to trisomy of chromosome 21. In particular, a deficit of growth and maturation of the brain is consistently described, and patients develop a variable degree of cognitive impairment, usually leading to dementia after 50 years of age [17]. Neuropathologically, severe neuronal loss has been described in the hippocampal formation, neocortex and in subcortical areas [17]. The formation of NFT and amyloid deposits occurs prior to neuro nal loss. Neurofibrillary degeneration with tau accumulation appears later in life. The hippocampal formation, including the entorhinal cortex, contains the highest number of NFT. Downs syndrome brain extracts contain significant amounts of insoluble tau[1,17].
Niemann-Pick Type C (NPC) Disease. This is a cholesterol storage disease with defects of intracellular trafficking of exogenous cholesterol derived from low density lipoproteins. Niemann-Pick type C includes juvenile dystonic lipidosis, ophthalmoplegic lipidosis, neuro visceral storage disease 6 with vertical supranuclear ophthal moplegia, and juvenile NPC disease [18]. The onset may be in infancy, early childhood, adolescence, or occasionally, in adulthood. Common neurological manifestations are clumsiness, ataxia, supranuclear gaze paresis, seizures and psychomotor retardation [19]. Brains from NPC patients exhibit neuronal distension in the cortex and swollen axons in the brainstem. In cases with a chronic progressive course, tau containing NFT are present in many parts of the brain, including the hippocampus, neocortex and several subcortical structures [20].