

EXOGENOUS AND ENDOGENOUS RISK FACTORS FOR
MATERNAL NON-DISJUNCTION OF CHROMOSOME 21
Sperling K1,* Pelz J2
*Corresponding Author: Professor Dr. Karl Sperling, Charité, Universitaetsmedizin Berlin, Institut fuer Humangenetik, Augustenburger Platz 1, 13353 Berlin, Germany; Tel.: +49-30-450-66081; Fax: +49-30-45-66904; E-mail: karl.sperling@charite.de
page: 5
|
|
INTRODUCTION
The basic principles of chromosomal segregation during mitosis and meiosis have been highly conserved during evolution and thus follow almost the same pattern in all eukaryotes. An interesting phenomenon can be seen by light microscopy of living cells: at the transition between prophase and metaphase the chromosomes show an oscillatory movement, and finally, all kinetochores are aligned in the equatorial plate. They are arrested there for a few seconds, and then anaphase begins abruptly, all chromatids/chromosomes separate synchronously and move towards the poles.
The brief arrest is the visible expression of a very efficient checkpoint control at metaphase. Displacement of a single chromosome from the equator, or attachment of a mitotic chromosome to only one half of the spindle prevents anaphase and thus reduces chromosomal non-disjunction to a minimum [1,2].
A large number of proteins involved in these processes have been identified, and direct proof exists in lower organisms (e.g., Saccharomyces, C. elegans, Drosophila) that mutations in the underlying genes affect chromosomal segregation and consequently, lead to a significant increase of chromosomal non-disjunction [3,4].
In man, in contrast to almost all other eukaryotes, non-disjunction is not a rare event (Fig. 1), affecting perhaps as many as 20 to 30% of all zygotes [5-8]. The majority of these cases is caused by non-disjunction during oogenesis which can be due to different mechanisms (Table 1). There is increasing evidence that maternal meiosis is an error- prone process due to the lack of a chromosome-mediated checkpoint control [9]. It is therefore logical to assume that this process is also sensitive to the effect of endogenous and exogenous factors.
Meiosis in the Human Female. Meiosis in the human female starts in the 3rd month of fetal life, when each ovary contains several million oocytes [Fig. 2(a)]. By the 6th month of fetal life, the oocytes reach the dictyotene stage of prophase I, in which they remain arrested until sexual maturity. However, most of the oocytes undergo programmed cell death (apoptosis). At puberty, their number is less than 200,000. Of these, only about 400 become ovulated. Thus, an oocyte which is ovulated by a 40-year-old woman was arrested at dictyotene for about 40 years and then completes the first meiotic division. The second meiotic division is not finished until after fertilization. In other words, the first and second meiotic division with its high rate of non-disjunction, takes place around conception [Fig. 2(b)]. At least from a theoretical point of view, chromosomal segregation should be most sensitive to environmental factors at this time, i.e., about 2 weeks after the last menstruation period.
Endogenous Risk Factors for Trisomy 21. Downs syndrome (DS) is the most common birth defect known to be associated with an abnormal karyotype. Most trisomy 21 is due to an error in maternal meiosis, whereby about 70% originate during meiosis I (MI) and about 20% during meiosis II (MII); a defective paternal meiosis is found in up to 8% of all cases [10,11]. More than 95% of all cases are due to free trisomy 21, only a small proportion arises by a mitotic error after fertilization. Both aspects, a conspicuous phenotype and a high proportion of new mutants, make surveillance of trisomy 21 particularly suitable for assessing mutagenic hazards and endogenous risk factors. Therefore, trisomy 21 is registered in nearly all monitoring programs for congenital malformations as a paradigm for aneuploid mutations. Since trisomy 21 has also been extensively analyzed at the molecular level, this article will focus on risk factors for trisomy 21. Its aim is not to give a comprehensive review but a brief, exemplary selection (Table 2).
Trisomy 21 is associated with advanced maternal age, which is the established and most significant risk factor for its occurrence. A minor, generally accepted, risk factor is the birth of a previous child similarly affected with free trisomy 21. These cases might be due to gonadal mosaicism or premature centromere division (PCD). However, it has been suggested that in some families with multiple trisomy 21 conceptions, a genetic predisposition to non-disjunction 21 may have been unmasked.
Evidence for genetic risk factors in meiotic non-disjunction are, amongst others, abnormal folate and methyl metabolism in mothers with DS infants, which are correlated with polymorphisms in the MTHFR and/or MTRR genes [12]. In other cases with recurrent aneuploidy, a role for mitochondrial DNA (mtDNA) mutations was suggested, which could also be relevant for the maternal age affect [13]. In addition, a number of gene mutations affecting the germ line or somatic cells have been identified that increase the risk of mitotic non-disjunction (Table 3).
We have studied a consanguineous Arab family with 10 children, of whom three had free trisomy 21. The parental karyotypes were normal and did not exhibit PCD. Based on 22 evenly distributed microsatellites, it was shown that the extra chromosome was of maternal origin, resulting from an error in MI (two cases) and MII. In addition, we found that the rate of somatic malsegregation of chromosome 21, but not of 18, 22 or X, was significantly increased in the mother. It was in the normal range in her mother, grandmother and, also in her three trisomic children who carry the same chromosomes 21. Thus, the high rate of maternal meiotic non-disjunction is paralleled by an increase of mitotic non-disjunction, however, confined to chromosome 21 [14]. The genetic basis of this phenomenon is obscure. It could be due to a recessive gene in combination with a specific chromosomal risk factor (Table 2).
There is clear evidence that in DS the recombination frequency of chromosome 21 was reduced for chromosomes with MI errors and was higher for MII errors, and both types were associated with advanced maternal age [15-18]. These altered levels of recombination represent associations, they are neither necessary nor sufficient causes for trisomy 21, since a number of control individuals arising by regular meioses (without non-disjunction) have recombinations in the same chromosomal areas as have their aneuploid counterparts, and the distribution of double exchanges are nearly the same for normal and aberrant meioses.
Exogenous Risk Factors for Trisomy 21. Numerous studies have tried to establish exogenous risk factors for trisomy 21 [reviewed in 5,10,19-21] (Table 2). Maternal age distribution and selective abortion after prenatal diagnosis have the strongest influence on its frequency in western countries. If these variables remain constant, then any sudden increase in frequency must be due either to chance or to an environmental factor. Nonetheless, even despite decades of research, apart from maternal age, no single exogenous factor responsible for trisomy 21 has been unambiguously identified.
Two convincing correlations were reported to explain two local clusters of trisomy 21 by hazards occurring around the time of conception: the ingestion of a chemical employed against fish parasites [22] and the inhalation of iodine-131 from the Chernobyl reactor accident [23]. There are two other epidemiological studies in which parents were exposed to a high dose of irradiation at the time of conception. These concerned the inhabitants of Kerala, India [24], and Yangjiang Province, China [25], who received high background radiation from monazite soil. The only relevant health problem was a significant increase in trisomy 21. Altogether, these studies imply that chromosomal segregation during human oogenesis around conception can easily be disturbed. Perhaps this is one explanation, apart from reporting bias, that a number of correlations between exogenous and endogenous factors and trisomy 21 were reported but could not be confirmed in other studies.
Table 1. Proposed mechanisms for non-disjunction
at meiosis.
|
1. True non-disjunction |
Both homologues segregate together |
|
2. Achiasmatic non-disjunction |
None or prematurely resolved chiasmata |
|
3. Premature separation of sister chromatids |
Segregation of a whole chromosome and a chromatid |
|
4. Anaphase lagging |
Loss of one chromosome (chromatid) |
|
5. Secondary non-disjuntion |
Somatic non-disjunction in maternal germ cells |
Table 2. Postulated risk factors of meiotic non-
disjunction in man.
|
Somatic mutations:
- defective MAD2 gene (OMIM 601467)
- defective BUB1 gene (OMIM 602452)
|
|
Germline mutations:
- Apple-Peel syndrome (OMIM 243605)
- Mosaic variegated aneuploidy syndrome (MVAS; OMIM 257300)
- MVA with total premature chromatid separation (OMIM 176430)
- Roberts syndrome (OMIM 268300)
- RECQ4-deficiency (Rothmund-Thomson S.; OMIM 268400)
|
|
Increasing maternal age:
- limited oocyte pool
- two-hit model susceptible bivalent
- abnormal processing of metaphase I
- defective spindle formation
- defective checkpoint control
|
|
Monogenic risk factors:
- defective folate metabolism
- apolipoprotein e4 allele
- Presenilin-1 gene polymorphism
- impaired function of mitochondria
- consanguinity (homozygosity for specific recessive genes)
|
|
Chromosomal risk factors:
- size of chromosomes
- NOR variants
- variation in alphoid DNA
- aberrant centromere structure
- premature centromere division
|
|
Environmental risk factors:
- parental irradiation
- oral contraceptives
- fertility drugs
- thyroid antibodies
- alcohol consumption
- viral infection
- ingestion of metriphonate
|
|
Others:
- reproductive activity
- seasonal variation in endocrine factors
|
Table 3. Selection of genes affecting mitotic non-
disjunction in man.
|
Somatic mutations:
- defective MAD2 gene (OMIM 601467)
- defective BUB1 gene (OMIM 602452)
|
|
Germline mutations:
- Apple-Peel syndrome (OMIM 243605)
- Mosaic variegated aneuploidy syndrome (MVAS; OMIM 257300)
- MVA with total premature chromatid separation (OMIM 176430)
- Roberts syndrome (OMIM 268300)
- RECQ4-deficiency (Rothmund-Thomson S.; OMIM 268400)
|

Figure 1. Incidence of aneuploidies in man.
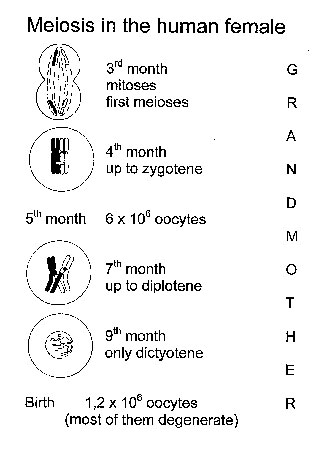
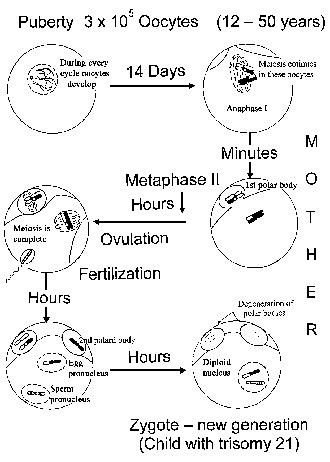
Figure 2. Meiosis in the human female. a) Meiosis starts in early fetal life and at birth all oocytes are arrested at the dictyotene stage. Most of the oocytes degenerate. b) Meiosis continues in individual oocytes after puberty. The first meiotic division occurs immediately before fertilization, the second a few hours thereafter (after [28]). In case of a zygote with trisomy 21, environmental effects during early oocyte development occurred in the grandmother, while effects during meiotic divisions operate in the mother.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|