

NOVEL MUTATIONS AND HAPLOTYPE ANALYSIS
OF GENOMIC POLYMORPHISMS OF GJB2 AND
GJB3 GENES ASSOCIATED WITH PROFOUND AND
MODERATELY SEVERE HEARING LOSS IN
PATIENTS FROM BASHKORTOSTAN
Dzhemileva LU*, Khidiyatova IM, Khabibullin RM, Khusnutdinova EK
*Corresponding Author: Dr. Lilya U. Dzhemileva, Institute of Biochemistry and Genetics, Russian Academy of Sciences, Prospect Octyabrya 69, 450054, Ufa, Russia; Tel: +07-3472-355255; Fax: +07-3472-356100; E-mail: Dzhemilev@anrb.ru
page: 41
|
|
RESULTS AND DISCUSSION
The 35delG mutation was detected in 35 of the 58 families (60%) with prelingual non-syndromic hearing loss. Twenty-three patients were homozygous (79.3%) and six heterozygous (20.7%). Thus, the 35delG mutation was observed on 52 out of 116 chromosomes (44.8%). The heterozygous patients were probably compound heterozygotes, carrying other GJB2 mutations along with the 35 delG mutation. Zelante et al. [8] have recently shown a very high frequency (~50%) of the 35delG mutation in Spanish, Italian and Israeli deaf patients. Therefore, these results suggested that the 35delG mutation underlay half of the cases of prelingual autosomal-recessive deafness. Also, more than 40 other mutations have been reported in the GJB2 gene.
In addition to the 35delG mutation, five different GJB2 mutations, including three novel mutations, were identified in our samples. In a child with a family history of profound non-syndromic hearing loss, we found a novel mutation in the GJB2 gene. The child carried the 35delG mutation and a new, transversion mutation, 254C®A, that caused the substitution of a serine residue for a tyrosine at codon 86, and impaired the transmembrane portion (M2) of the protein. The second mutation was 235delC. This deletion resulted in a frameshift converting codon 78 from leucine to cysteine, followed by a stop codon at position 79. The third and fourth (novel) mutations were 313/314 delAA and 314delA. These mutations cause a frameshift at codon 111, resulting in a stop codon at positions 112 and 117, respectively. The mutation 360delG was found in only one family. This deletion caused a frameshift, that resulted in chain termination after addition of novel amino acids. Furthermore, we also identified a novel mutation, 557A®T, in the GJB3 gene. This mutation was detected in 5% of all mutant chromosomes with sensorineural hearing loss. Interestingly, the audiogram of these patients showed a bilateral mild sensorineural hearing loss at all frequencies, mainly at high frequencies. The transversion 557A®T, resulted in the substitution of an isoleucine residue for a phenylalanine at codon 186, and impaired the trans-membrane portion (M3) of the protein. It was associated with dominant high-frequency hearing loss.
We could not detect any mutation in the alleles of the GJB2 and GJB3 genes on 50% of the studied chromosomes. Some studies support the hypothesis that other factors may modify the phenotypic effects of mutations in the GJB2 gene, as the existence of modifier genes have been known for a long time. A nuclear-mitochondrial gene interaction has also been proposed. In most cases, the genetic counseling of heterozygous deaf individuals remains a dilemma. One possibility is that in these individuals the hearing loss is due to a mutation in an entirely different gene.
In order to explore the ancestry of GJB2 mutations in the patients with non-syndromic inherited deafness, we estimated linkage disequilibrium between the 35delG mutation, other mutations, and flanking microsatellite markers D13S143, D13S292, and D13S175. For evaluation of the allele frequencies for the D13S143, D13S292, and D13S175 loci, we performed genotyping on 30 affected, unrelated families with the 35delG mutation and other mutations (254C®A, 235delC, 313/314delAA, 314delA, 360delG) in the GJB2 gene from Bashkortostan. Allele frequencies of the three polymorphic loci, D13S143, D13S292 and D13S175, are summarized in Table 1.
Both among patients and in the control group, linkage disequilibrium values were significant (p <0.0001) between alleles with the 35delG mutation and alleles of markers D13S143, D13S292 and D13S175, respectively (Table 1). The 35delG mutation was in significant linkage disequilibrium with allele 13 of D13S143, which is most unusual among healthy people (DSt13 = 0.6304) (Table 1). All the affected individuals with the 35delG mutation showed high homozygosity for some alleles of markers D13S143, D13S292 and D13S175 (observed heterozygosity was 0.36, 0.39 and 0.25, respectively). Assortative mating among the deaf, however, could give an explanation to this extremely high homozygosity. Haplotypes were constructed using the three genetic markers D13S143, D13S292 and D13S175. The haplotype diversities in the studied groups are given in Fig. 2; the chromosomes with the 35delG mutation have the lowest haplotype diversity (0.6875) and the mutant chromosomes without the 35delG mutation, the highest (0.9632).
Mutant chromosomes without the 35delG mutation carried 18 different haplotypes on 13q11-13q12. Haplotype 13-18-15 was observed on 10% of all mutant chromosomes without the 35delG mutation, and haplotypes 13-20-17 and 13-17-16 were detected on 10% chromosomes with the 35delG mutation. The frequency variations in various haplotypes among the studied groups of chromosomes were statistically significant [c2 = 152.2; p <0.0001].
Connexin 26 is a member of a large family of gap junction membrane proteins that mediate electrical and metabolic coupling between adjacent cells. Several findings support the importance of connexin 26 in auditory transduction. Immunohistochemical and structural analysis of the connexin 26 protein in the rat cochlea suggests that gap junctions in epithelial and connective tissue cells are involved in recycling endolymphatic potassium ions through non-sensory cells during the transduction of voltage gating in these channels [20,21].
Given the high frequency of GJB2 mutations among deaf children from central and northern Europe, the testing for the GJB2 gene among deaf children in Bashkortostan is worthwhile in order to provide diagnosis at an early age. Furthermore, unnecessary invasion could be avoided, since GJB2 mutations are not associated with syndromic hearing loss or with inner ear malformations [3,4].
In conclusion, our results indicate that in Bashkortostan, testing for disorders of the GJB2 gene should be the first step in determining the cause of hearing loss. The screening for mutations in the GJB2 gene, especially the 35delG mutation, should be a routine investigation of either familial or sporadically affected individuals with congenital or prelingual hearing impairment.
Haplotype analysis of the markers D13S292, D13S175 and D13S143 in the peri-centromeric region of chromosome 13 helped to optimize DNA diagnosis of non-syndromic deafness among patients from Bashkortostan. Prevalence of the 35delG mutation in Bashkortostan may indicate a founder effect. The results presented here can be used for the development of a simple molecular test that is sure to be of considerable help. The molecular diagnosis is essential for genetic counseling, and may also allow early rehabilitation and a better development of speech and language of affected children in these families.
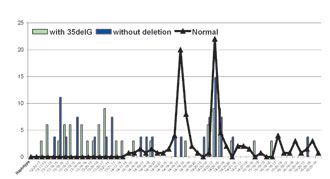
Figure 2. The haplotype diversities in the studied groups.
Locus Allele |
Chromosomes |
|
(repeat #) |
With 35delG (n = 57)
p |
Without 35delG (n = 59)
p |
Normal (n = 218)
p |
DSt |
D13S143
1(12)
2(13)
3(14)
4(15)
5(16)
6(17)
7(18) |
0.0175±0.014
0.5789±0.065
0.3684±0.063
0.3508±0.061
0
0
0 |
0.0338±0.025
0.5593±0.064
0.3389v0.061
0.0508±0.028
0.0169±0.016
0
0 |
0
0
0.4266±0.033
0.4495±0.033
0.0871±0.019
0.0321±0.011
0.0045±0.0035 |
0.1143
0.6304
0.075
0.4716
0.2160
0.1277
0.047 |
D13S292
1(17)
2(18)
3(19)
4(20)
5(21) |
0.052±0.029
0.263±0.058
0.157±0.048
0.438±0.065
0.087±0.037 |
0.0847±0.036
0.2203±0.053
0.2881±0.052
0.3389±0.061
0.0677±0.032 |
0.009±0.006
0.050±0.014
0.403±0.033
0.454±0.022
0.082±0.018 |
0.1544
0.2704
0.1934
0.067
0.009 |
D13S175
1(13)
2(14)
3(15)
4(16)
5(17)
6(18)
7(19)
8(20)
9(21) |
0
0
0.2105±0.053
0.2105±0.053
0.3859±0.060
0.1754±0.050
0
0.0175±0.011
0 |
0.0338±0.025
0.0677±0.032
0.3389±0.061
0.1864±0.050
0.2203±0.053
0.1355±0.044
0
0.0169±0.016
0 |
0
0
0.0275±0.011
0.1422±0.023
0.4862±0.036
0.2155±0.027
0.1009±0.020
0.0229±0.012
0.0045±0.043 |
0.093
0.1293
0.3477
0.074
0.1888
0.0777
0.2305
0.0200
0.4787 |
|
|
|
|

 |
Number 28
Vol 28 2025 Supplement |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|
|