

PRADER-WILLI AND ANGELMAN SYNDROMES
IN THE GREEK POPULATION: A CLINICAL AND
MOLECULAR STUDY
Salavoura K*, Sofocleous C, Mavrou A, Kalaitzidaki M,
Frysira H, Kolialexi A, Zafiriou D, Yapijakis C, Metaxotou E
*Corresponding Author: : Katerina Salavoura, MD, PhD, Department of Immunology and Histocompatibility, Aghia Sophia Childrens Hospital, Thivon and Papadiamantopoulou, Athens, Greece; Tel.: +30-210-7467766; Fax: +30-210-7757401; E-mail: Salavoura_Katerina@hotmail.com
page: 27
|
|
RESULTS
Clinical findings
Based on published criteria [1,5], clinical examination classified 40/114 (35%) PWS and 32/57 (56%) AS patients as having typical phenotype of the syndrome. Typical PWS patients presented with hypotonia in the neonatal period (88%), obesity after the 3rd-5th year (84%), developmental delay (85%), characteristic face (78%) and small hands and feet (68%) (Table 1). The clinical presentation of AS was characterized by severe developmental delay (91%), lack of speech (84%), ataxic gait (69%), spasms (63%) and improper laughter (63%) (Table 2).
In 5 of 15 children referred with hypotonia in the neonatal period, the diagnosis of PWS was confirmed, since DNRP analysis revealed deletion or uniparental disomy in the 15q11 region. The remaining ten children were re-examined 5 years later and the diagnoses were as follows: 1 Robinow syndrome, 1 Rett syndrome, 1 central core disease, 1 agenesis of corpus callosum, 1 tetraplegia due to birth injury, 1 developmental delay of unidentified reason. Four children had normal development.
Cytogenetic investigation
Cytogenetic analysis revealed a normal karyotype in 59 patients, a visible cytogenetic deletion in the region 15q11-13 in 3 cases and a Robertsonian translocation 45 XY, t (15; 15) in 1 case.
Molecular investigation and fluorescent in situ hybridisation analysis
Molecular investigation of patients using PCR analysis confirmed the diagnosis of PWS or AS with the detection of absent paternal or maternal alleles, respectively. Schematic presentations of molecular results are shown in Figures 1 and 2. Diagnosis was confirmed in 39/114 of referrals for PWS (34%) and revealed 28 deletions (72%) and 11 cases with uniparental disomy (28%). Uniparental disomy was observed also in the patient with the 45XY, t (15; 15) karyotype. Most of the typical PWS patients (28/40) had a molecular alteration (70%), while molecular findings were also observed in 11% of atypical cases (7/62).
Table 1. Laboratory findings in Prader Willi and Angelman syndrome patients.
|
|
PWS Patients |
% |
AS Patients |
% |
|
Referred cases |
114 |
|
57 |
|
|
Males |
79 |
|
22 |
|
|
Females |
35 |
|
35 |
|
|
Mean age (years) |
5.5 |
|
7.6 |
|
|
Karyotype abnormalities |
4 |
3.5 |
0 |
0.0 |
|
FISH analysis |
23 |
20.0 |
11 |
19.0 |
|
Abnormalities detected by FISH |
1 |
0.8 |
0 |
0.0 |
|
Molecular analysis |
114 |
|
57 |
|
|
Molecular changes |
39 |
34.0 |
18 |
32.0 |
|
Confirmed diagnosis |
40 |
35.0 |
18 |
32.0 |
Table 2. Clinical characteristics of 40 patients with a typical Prader Willi syndrome (PWS) phenotype and 32 patients with a typical Angelman syndrome (AS) phenotype.
|
Clinical Characteristics |
PWS Patients
(n = 40) |
% |
AS Patients
(n = 32) |
% |
|
Gestation period |
Normal 35 |
88.0 |
Normal 23 |
72.0 |
|
Delivery |
Cesarian 12 |
35.0 |
Cesarian 5 |
16.0 |
|
Mean birth weight |
2.940 gr |
|
3.110 gr |
|
|
Hypotonia in the neonatal period |
35 |
88.0 |
10 |
31.0 |
|
Feeding problems |
26 |
65.0 |
N.E. |
N.E. |
|
Obesity after the 3rd-5th year |
27/32 |
84.0 |
N.E. |
N.E. |
|
Characteristic face |
31 |
78.0 |
18 |
56.0 |
|
Developmental delay |
34 |
85.0 |
32 |
100.0 |
|
Short stature |
13 |
33.0 |
N.E. |
N.E. |
|
Small hands and feet |
27 |
68.0 |
N.E. |
N.E. |
|
Hypognadism |
15 |
35.0 |
N.E. |
N.E. |
|
Ataxic gait |
N.E. |
N.E. |
22 |
69.0 |
|
Improper laughter |
N.E. |
N.E. |
20 |
63.0 |
|
Lack of speech |
N.E. |
N.E. |
27 |
84.0 |
|
Spasms |
N.E. |
N.E. |
20 |
63.0 |
|
Microcephaly |
N.E. |
N.E. |
20 |
56.0 |
|
Behavioral problems |
23 |
58.0 |
N.E. |
N.E. |
Table 3. Correlation between phenotype and genotype in Prader Willi (PWS) and Angelman syndrome (AS) patients.
|
|
n |
Molecular
Confirmationa |
% |
Uninformative
or Normal |
% |
|
PWS Patients
- Typical presentation
- Non typical presentation
|
114
40
74 |
28/40
11/74 |
70.0
15.0 |
12/40
63/74 |
30.0
85.0 |
|
AS Patients
- Typical presentation
- Non typical presentation
|
57
32
25 |
18/32
0/25 |
56.0
0.0 |
14/32
25/25 |
44.0
100.0 |
a Includes deletions, UPD.
Diagnosis was confirmed in 18/57 of referrals for AS examination (32%) and all patients had deletions. Deletions were observed in 56% of the patients with typical AS presentation. No deletions were found in patients with atypical presentation.
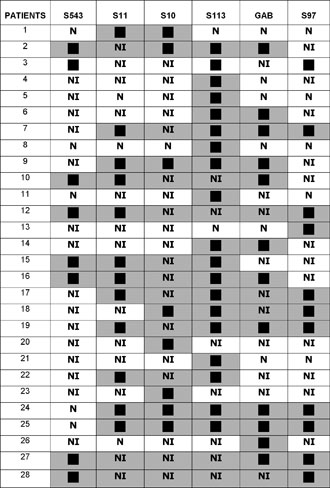
Figure 1. Schematic presentation of deletions in PWS patients.
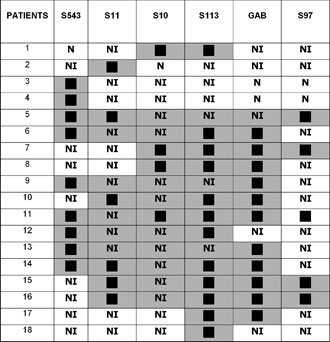
Figure 2. Schematic presentation of deletions in AS patients.
PWS patients No 118 and No 119, identical twins with typical clinical presentation, had a large deletion encompassing the whole 15q11-13 region except the S543 locus. Patients No 77 and No 78 were also identical twins with typical AS and deletions limited to the S543 locus.
FISH analysis using the SNRPN probe revealed two hybridization signals in 33 cases, which were not informative by PCR analysis, including the case of 45XY, t (15; 15). In 1 patient in whom molecular investigation was not informative, only one signal was observed, indicating a deletion for the SNRPN gene.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|